"What Happened to Blog Syn?"*
I get asked that question a few times every week. With questions about scientific (ir)reproducibility and data integrity popping up everywhere, it would seem apropos to resurrect the blog from its months-long slumber. So why aren't we posting?
Changing Circumstances: Five synthetic chemists ran most of the experiments for the first three posts.
Where are they now?
- One moved to another country, and hasn't started in his new lab
- One defended his Ph.D., and moved to another state
- One passed his qualifying exams, and wrote a bunch of papers for a new lab
- One faced a daunting funding situation at his small company, along with changes in work responsibilities
- (I don't know where the last person is...!)
So . . . life happened.
Time: The process of coordinating and writing a Blog Syn entry takes a while. Let's compare:
Just Like Cooking: Read interesting paper, dig up literature, make some graphics, edit, post.
Time: 20-60 minutes
Blog Syn: Read interesting paper, discuss with Twitter community and coworkers, read supporting literature and SI, coordinate across multiple time zones by email, order reagents, run experiments, purify, analyze, compare data, coordinate response by email / Skype / Twitter, construct post, peer edit, post.
Time: 2-4 weeks
Resources: We have no formal funding model. At present, our labs bear Blog Syn's costs, and that's OK for reagents you have just sitting around. It becomes dicey, though, to convince purchasing managers to order unusual reagents for your pet projects.
People: Despite multiple attempts to recruit chemists to the cause, we've not added anyone new to our roster. I've sent out at least three separate email blasts, had some nibbles, but no one has committed. I feel it would be disingenuous to run each experiment in triplicate myself, since that only proves that I could repeat all my own mistakes.
I think what's happening to Blog Syn is what happens to all volunteer organizations over time. Life happens. People move. Interests shift and change. Resources dwindle. The message and mission grow muddy.
*HELP WANTED: I'd really like to keep this going; I have preps picked out to try, folks to review and publicize posts, even some potential funding to fall back on. So, who's with me? Who wants to help revitalize Blog Syn?
If you've been dragging your heels and wish to get involved, don't hesitate to contact me!
Operators are standing by...
Thursday, July 18, 2013
Sunday, March 3, 2013
Blog Syn #003A - Secret Ingredient
(Disclaimer: The following experiments do not constitute rigorous peer review, but rather illustrate typical yields obtained and observations gleaned by trained synthetic chemists attempting to reproduce literature procedures. We've taken efforts to stay close to the original procedure, using similar glassware, equipment, and reagents wherever possible. Images have been cropped and scaled to fit in the allotted space, but have not been digitally altered otherwise.)


It's been a wild month since our post concerning IBX-promoted benzylic oxidation. This piece provoked energetic discussion, evidenced by >200 comments (and counting!) between Blog Syn, two Pipeline posts, and Rich Apodaca's insightful discussion at Depth First.
Before we wrap up this topic, we at Blog Syn first wish to thank Prof. Phil Baran, Dr. Tamsyn Montagnon, and Dr. Yong-Li Zhong (the original authors) for engaging us honestly and professionally, and helping uncover some of the hidden factors that make this reaction work.
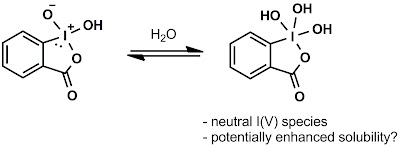
I think we now know the most important one: water.
After reading Tamsyn and Phil's submissions to #003, we were puzzled: what was so different about our reaction conditions? Everyone was following the same procedure [Santagostino, J. Org. Chem. 1999, 64(12), 4537] to make their IBX...right?
Well, not really. No one's published the actual NMR spectrum for IBX in quite some time. Neither this paper, nor Kirsch OL 2006, nor JOC 1999[1], nor Tet. Lett. 1994 (which the JOC refers back to) do much more than provide a list of peaks. This next image shows the NMR spectra provided by Phil, Tamsyn, and by an anonymous commenter online at Blog Syn. Note the rather large water peak at 3.4 ppm (possibly from some pre-opened d6-DMSO?)
Next, I made some fresh IBX strictly following the JOC 1999 paper, which includes two acetone washes to dry the IBX. The small bump at 8.4 ppm, which we had believed to be an impurity, might actually be the OH proton of non-solvated IBX. Note that, as I titrate in additional H2O the bump disappears, the aromatics shift and coalesce, and the 'signature' water peak grows in at 3.4 ppm!
After discussion with the Blog Syn crew and the authors, we propose "hydrated IBX" may be the active species for the oxidation:
Co-author Yong-Li suggested some 18O-labeled experiments to determine water's role in the reaction. Sadly, we're not equipped for this type of chemistry, but if any readers are, please feel free to report your findings!
 |
| Rxn, 20 h, w/H2O |
Now the fun part: "hydrated IBX" performs the oxidation!
Here's a (straight-baseline) TLC; runs "A" and "B" have varying [H2O] in the solvent, which does seem to accelerate the reaction (right). Prof. Nicolaou had remarked on this trend in the Details of ACIEE 2002, 993 (see p. 996) noting that enone dehydrogenation yields decreased when solvent was pre-dried over 4A sieves.
Here's a (straight-baseline) TLC; runs "A" and "B" have varying [H2O] in the solvent, which does seem to accelerate the reaction (right). Prof. Nicolaou had remarked on this trend in the Details of ACIEE 2002, 993 (see p. 996) noting that enone dehydrogenation yields decreased when solvent was pre-dried over 4A sieves.
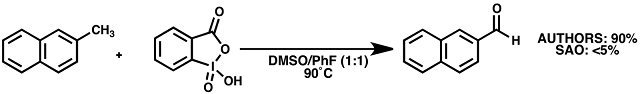
Using "wet" DMSO and fluorobenzene (5:1), and heating to 95 deg C in a foil-coated, fully-submerged screwtop vial, I'm now able to convert 2-methylnaphthalene to naphthaldehyde in 52% yield (brsm). My colleagues have also observed increased conversion of toluene and 4-bromotoluene, but neither go to completion by the published time points.
So, what should we recommend? It seems clear that several critical details are missing from the original Supporting Info. How much have we deviated from the initial conditions?
Based on the information above, and the results in our hands over multiple runs, we will change our recommendation to Reproducible with optimization.
We look forward to future communication with all authors involved, and hope that this interchange inspires chemists to have another look at IBX-promoted oxidation reactions...just add water!
[1] We should point out that JOC 1999 does include KF titration values, but the low water % detected does not rule out sufficient hydration of some IBX. Note also that the elemental analysis values deviate enough to include trace water.
Monday, February 18, 2013
Blog Syn #003: Benzylic Oxidation of Arylmethanes by IBX
(Disclaimer: The following experiments do not constitute rigorous peer review, but rather illustrate typical yields obtained and observations gleaned by trained synthetic chemists attempting to reproduce literature procedures. We've taken efforts to stay close to the original procedure, using similar glassware, equipment, and reagents wherever possible. Images have been cropped and scaled to fit in the allotted space, but have not been digitally altered otherwise.)
Update 2: 2/27/13 - Added Tamsyn NMR and email
Update 1: 2/25/13 - Added Phil NMR and email

Well, let’s see how things stack up for BRSM and See Arr Oh.
Author Response: Sent over by Prof. Phil Baran, Feb 10, 2013 (highlights mine):
"...my recollection was that the IBX we made was ALWAYS completely soluble in DMSO at higher temperature (80 degrees I recall), and dissolved in DMSO prior to use in the oxidation reaction.. I always tried to do my reactions in pure DMSO. In addition, we never ordered IBX, it was always made freshly before use, ensuring that it was made with an internal heating temperature of 73 degrees Celsius to get the optimal yield and purity of product. IBX quality was discerned by 1H NMR in DMSO prior to use. I'd recommend you try these reactions without the fluorobenzene, which was used with certain substrates that were not soluble in pure DMSO at elevated temperature.
B.R.S.M. (again) - We decided to re-run the reaction, given Prof. Baran's helpful suggestions.
(Update: 2/21/13) See Arr Oh here again: noting suggestions made by Prof. Baran and various commenters, I re-set a 1.0 mmol reaction, this time in pure DMSO with 0.1 volume of H2O under a lab air environment. I observed the material going mostly into solution (60 degrees), and a lemon-yellow color evolved inside of 2 hours.
*See Arr Oh here: We at Blog Syn thank Prof. Baran for his willingness to re-run the reaction, and for sending over visual evidence. Once we have a chance to run it in our hands with the improved conditions, we will reconsider the above recommendation.
Author Addendum II: Dr. Tamsyn Montagnon has also written in with a duplicated reaction. Below, I've reproduced her email and pictures in their entirety:
"You may want to consider the following:
*We at Blog Syn thank Dr. Montagnon for her willingness to duplicate the reaction and for sending over visual evidence. We will incorporate her reaction into our final assessment.
Update 2: 2/27/13 - Added Tamsyn NMR and email
Update 1: 2/25/13 - Added Phil NMR and email
Ref: Nicolaou, K. C.; Montagnon, T.; Baran, P. S.; Zhong, Y. L. J. Am. Chem. Soc. 2002, 124, 2245–2258.
Supporting Info
Supporting Info
Experimenters: Three - B.R.S.M. and coworker (United Kingdom), See Arr Oh (Somewhere, U.S.A.)
Recommendation: Pending Blog Syn repeat
Initial - Difficult to reproduce - little / no product observed or isolated.
*See below for Phil Baran's response and repeated reaction.
Initial - Difficult to reproduce - little / no product observed or isolated.
*See below for Phil Baran's response and repeated reaction.

In Blog Syn's third installment, we examine a benzylic oxidation reaction pointed out by Chemjobber commenter Jose. Jose claims “0% conversion for multiple substrates with multiple chemists" using this reaction. The source paper is one of a four-part series penned by K.C. Nicolaou and Phil Baran describing the multitudinous uses of o-iodoxybenzoic acid (IBX) in organic synthesis. With Jose’s comment fully in mind, the Blog Syn Team members See Arr Oh and BRSM put the reaction to the test.
A day in the library can save you a week in the lab, so let’s examine this paper’s impact using SciFinder: it's been cited 179 times from 2002-2013. Using the “Get Reactions" tool, coupled with SciFinder’s convenient new “Group by Transformation” feature, we identified 54 reactions from the citing articles that can be classified as “Oxidations of Arylmethanes to Aldehydes/Ketones" (the original reaction's designation). Of these 54 reactions, only four (4) use the conditions reported in this paper, and all four of those come from one article: Binder, J. T.; Kirsch, S. F. Org. Lett. 2006, 8, 2151–2153, which describes IBX as “an excellent reagent for the selective oxidation to generate synthetically useful 5-formylpyrroles.” Kirsch's yields range from 53-79% for relatively complex substrates, not too shabby.
Well, let’s see how things stack up for BRSM and See Arr Oh.
TRIAL 1 / 1a:
Experimenter: BRSM (and friend!)
Scale: 1 mmol (the paper claims 0.1 mmol to 1 mmol, without giving scales for specific entries. Used SI general procedure)
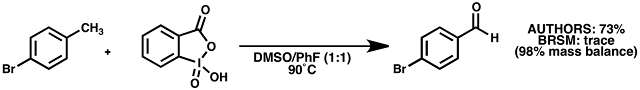
Example: 4-bromotoluene (table 5, entry 6)

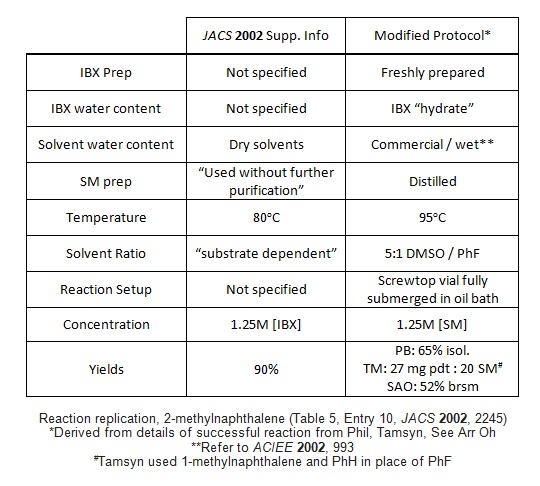
Reagents: Aldrich fluorobenzene, 99%, borrowed and used as recieved; Aldrich 4-bromotoluene, 98%, used as is; ACROS Extra Dry DMSO, 99.7% / AcroSeal'd bottle (almost new), IBX prepared according to JOC 1999, 4537, stored in freezer ~6 months.
Observations: A 1:1 mixture of DMSO:PhF was used at the suggested concentration of 1.25 M with respect to IBX as starting material wasn't soluble in DMSO. The reaction was carried out in a sealed tube that had been oven-dried for several days at 140 ºC and cooled under argon. IBX was found to be almost completely insoluble in this solvent system even at 90 ºC (Dry-Syn temperature). Stirred for 16 h, causing no apparent change to the white suspension. Worked up as described, except that saturated NaHCO3 solution was substituted for the 5% solution used by the authors. A white solid was recovered (177 mg). TLC after workup (20% ethyl acetate in petrol 40-60) showed a faint new spot at Rf 0.5 that stained rapidly with KMnO4 dip without heating.
Analysis: Crude 1H NMR showed trace product, with the balance being made up by starting material with traces of PhF and 2-iodobenzoic acid. As the mass recovery was good (177 mg crude back from 171 mg starting material) and the product aldehyde is not volatile (bp 240 ºC), it must be concluded that the reaction did not occur.
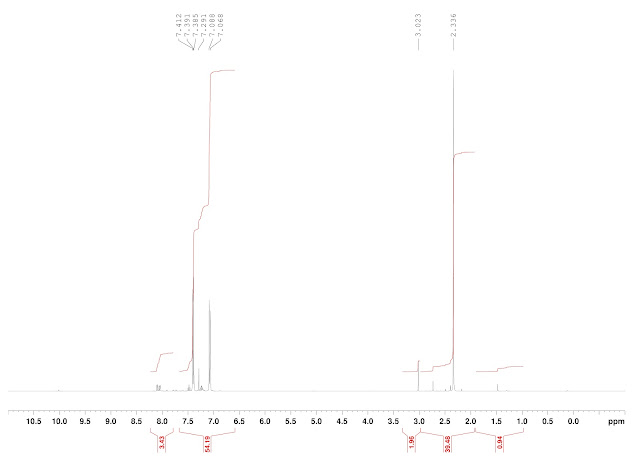 |
| BRSM, crude NMR |
Advice: As See Arr Oh and others have observed, the main problem with trying to repeat this work is the lack of an unambiguous Experimental procedure. Reactions were apparently carried out in either DMSO, or 1:2 DMSO:Fluorobenzene according to the paper, but the SI says that the ratio is substrate-dependent and that the reaction is faster if the amount of fluorobenzene is minimised. With such scant detail, it's pretty hard to say how precisely we repeated the reaction. Also, although I'm reasonably sure my IBX was good, I wonder what the authors used. As they don't state otherwise I presume that commercial IBX was used, if their general SI preamble is to be believed. I've never bought IBX, but I imagine it's packed out with lots of stabilisers, which might be important for the reaction. Would be interesting to try again with commercial IBX just to be sure.
Extra Credit: A lab-mate of mine tried a 1 mmol reaction on toluene (table 5, example 1), which was obtained from Fisher Scientific via our communal drying towers, using the same reagents and set-up as above. His tube was in the same Dry-Syn as mine, for the same time, and worked up identically. No colour change was observed and TLC against benzaldehyde seemed to indicate that no product had formed in the reaction before work-up. His crude NMR is a bit different to mine (presumably a lot of toluene was lost during workup and concentration), but still shows a serious lack of aldehydes.
 |
| "Lab friend," toluene, with IBX |
TRIAL 2:
Experimenter: See Arr Oh
Scale: 1.0 mmol
Example: 2-methylnaphthalene (Table 5, Entry 10)

Reagents: ACROS fluorobenzene, 99%, dried over 4A sieves; ACROS 2-methylnaphthalene, 97%, Alfa DMSO, 99.9% HPLC / ChemSeal bottle, IBX prepared in-house following JOC 1999, 4537, recrystallized, stored in freezer ~6 mos. IBX so prepared was active in enone dehydration reaction (see: Baran, Nicolaou ibid.)
Observations: Combined all reagents in oven-dried pressure tube under N2 stream. White suspension in 1:1 DMSO / fluorobenzene. Heated to 85-90˚C on sand bath. Suspension turns peach-orange, then deep yellow. Heated 19 h. TLC at 19 h shows faint spot at Rf 0.5 (5% MTBE / Hept). Workup: bicarb wash, extract with MTBE 3x, washed with H2O, dried over Na2SO4, concentrated to yellow solid, 220 mg.
Analysis: Crude 1H NMR: <5% product observed.
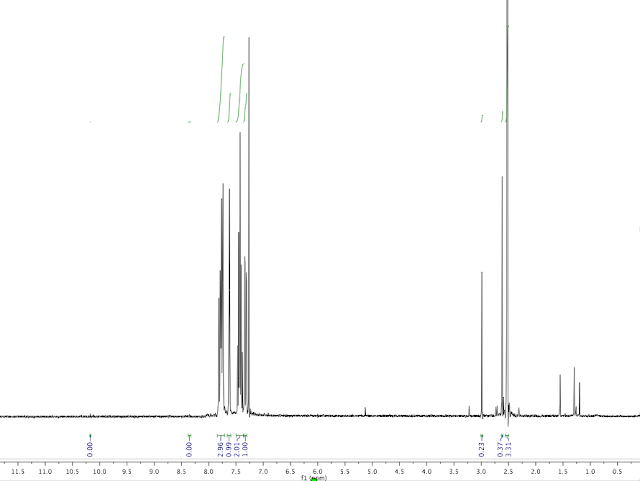 |
| SAO, Crude NMR |
{kind=link}
Advice: Supporting Information indicates a "substrate dependent ratio [of fluorobenzene:DMSO] with fluorobenzene conc. being minimized." My reaction never went completely into solution, despite attempts to sonicate / shake flask. It's possible that a different ratio of solvent may have promoted the reaction, but I'm skeptical.
 |
| SAO, Crude solid |
 |
| SAO, Unreacted mixture |
Author Response: Sent over by Prof. Phil Baran, Feb 10, 2013 (highlights mine):
"...my recollection was that the IBX we made was ALWAYS completely soluble in DMSO at higher temperature (80 degrees I recall), and dissolved in DMSO prior to use in the oxidation reaction.. I always tried to do my reactions in pure DMSO. In addition, we never ordered IBX, it was always made freshly before use, ensuring that it was made with an internal heating temperature of 73 degrees Celsius to get the optimal yield and purity of product. IBX quality was discerned by 1H NMR in DMSO prior to use. I'd recommend you try these reactions without the fluorobenzene, which was used with certain substrates that were not soluble in pure DMSO at elevated temperature.
It's unfortunate that the SI was not clear enough for you and your colleagues to repeat it, but I can assure you that this reaction IS reproducible and was done independently by several highly qualified chemists within the Nicolaou laboratory prior to publication. There might also be value in reproducing one of the substrates reported in one of the other literature examples that use IBX for benzylic oxidation (the 2006 Org. Lett., page 2151 you mention for example). In addition to the papers that use stoichiometric IBX others have found catalytic versions also capable of benzylic oxidation."
[Update(2/25/13) - Tamsyn Montagnon was indadvertently left off the original email chain.]
2/12/13
"It is right to suggest doing these reactions without fluorobenzene - I think a lot of the issue is that simple alkanes are pretty tricky in these reactions, because they aren't soluble in DMSO so you get little micelles floating on the surface and not reacting – ironically, this is a reaction that works better on more complicated substrates that have more functionality to assist in solubilizing them....FB certainly hinders the reaction (again probably a facet of creating almost a two phase reaction mixture)......and IBX is most certainly completely soluble in DMSO at RT, let alone elevated temps, anybody that has ever oxidized an alcohol with IBX knows this - IBA on the other hand is not, it crashes out during IBX oxidations and hinders the work of IBX (probably by being more Lewis acidic and sequestering non-productively substrate - if you add extra equivs of IBX once you have IBA in a reaction (in alcohol oxidations, dehydrogenations, benzylic oxidations whatever) you don't push it to completion you just end up with more IBA crashed out. The fact that they made a "Dess Martin" from their IBX means nothing ...God knows what oxidant you get if you heat up an IBX/IBA mixture in (Ac)2O."[Update(2/25/13) - Tamsyn Montagnon was indadvertently left off the original email chain.]
2/12/13
TRIAL 3:
B.R.S.M. (again) - We decided to re-run the reaction, given Prof. Baran's helpful suggestions.
Based on these comments, I decided that I'd have to remake the IBX fresh to their specifications in order to test the reaction properly. At first, I made IBX using the second procedure in the Oxone JOC paper (i.e. using 3 equiv. Oxone and a more dilute reaction mixture). I repeated the reaction on the scale (5g) and concentration given in the paper. On Phil's suggestion, I also used a thermometer and a great deal of time to ensure that the internal temperature was held at 70-73 degrees C. As promised, this batch contained no 2-iodobenzoic or 2-iodosobenzoic acid, but it did have around 5% of some other impurity I couldn't identify. The prep supposedly delivers 99% analytically pure IBX, so I was a little disappointed.
 |
| BRSM's new IBX batch |
It's interesting to note at this point that the NMR data for IBX given in the Org. Synth. prep for Dess-Martin seems a little odd; two of the aromatic protons have coupling constants of 14.8 Hz, which seems inexplicably large to me. The chemical shifts given in the Org. Synth. prep do match the literature, but I can't find coupling constants given anywhere else for IBX to compare. Mine are not that large. Strange. Also, all batches of IBX I have analyzed by NMR have a broad peak around 10, which I'd always put down the the OH, but this doesn't seem to be mentioned in the literature.
**This batch was totally soluble in neat DMSO at RT**
I checked my 2-iodobenzoic acid starting material by NMR - it was pure and free from impurities. I really wanted to make "perfect" IBX to give the benzylic oxidation a fair hearing, so I borrowed some 2-IBA and some Oxone from another group and tried again. I produced another batch, essentially identical to the first one, which is to say, ~95% pure by NMR. My colleague and I re-ran our benzylic oxidations on PhMe and 4-Br-PhMe with this "new" IBX. Based on the authors' recommendations we used neat DMSO. The IBX didn't dissolve immediately, although the literature (Tet. Lett.) says that it can take 10-15 mins stirring to go in. We starting heating straight after adding it and about 10 mins later, at an oil bath temp of 40 degrees C, we obtained clear solutions.
In the case of PhMe, the results were similar, with perhaps a larger aldehyde peak in the crude NMR, although given the volatility of the toluene we couldn't really measure conversion accurately. Benzaldehyde was also visible by TLC, which it wasn't for the first run. My colleague attempted a column but no aldehyde could be isolated. For the 4-Br-PhMe (which I ran) the results were quite different from last time. The mass recovery was worse - only 100 mg back from 171mg, compared to perfect recovery last time - although the same scale and workup were used. Also, the crude NMR was messier and didn't seem to contain as much unreacted SM. Still apparently <5% aldehyde, though. And the literature boiling point is ~240 degrees, so I doubt I lost it.
TRIAL 4:
(Update: 2/21/13) See Arr Oh here again: noting suggestions made by Prof. Baran and various commenters, I re-set a 1.0 mmol reaction, this time in pure DMSO with 0.1 volume of H2O under a lab air environment. I observed the material going mostly into solution (60 degrees), and a lemon-yellow color evolved inside of 2 hours.
Checking the reaction by TLC at 12 hours, however (see Just Like Cooking) did not indicate a much better result than I had obtained previously. Perhaps, as suggested, my IBX is simply too old; commenters' suggestions to re-make a new batch, recrystallize, or test IBX by iodometric titration are all under consideration.
I have heard that BRSM may be running the reaction with other co-solvents / conditions, so keep those fingers crossed!
*Author Addendum: One week after our initial post, Prof. Phil Baran wrote in with a successfully duplicated reaction and a much more elaborate experimental write-up. Below, I've reproduced his email and pictures in their entirety.
"I write regarding your entry on IBX oxidation of aryl methyl groups which your team found to be irreproducible. I have subsequently had a student run this reaction under the published conditions and the results, documented with pictures of everything, are below. I would like to mention that in the future it is very easy for you or any of your readers to contact me directly with any questions – those experiencing trouble could have emailed me anytime. Looking back on those days, well over a decade ago, I remember KC watching Yong-Li (one of the most incredible chemists I've ever worked with) and I do these reactions and us sharing TLC plates with him in our excitement. It was fun to do this reaction again and it brought back nice memories :)
Since your blog is quite new, I think it might be nice if in the future you give the authors a week or so to respond to claims of irreproducibility. After all, the paper has been out for over a decade so would an extra week hurt? You are, I suppose, in search of the truth. And the truth cannot always be realized with "Speed and Aplomb" as your blog advertises. Incidentally, my colleague Jin-Quan Yu has also reproduced his reaction and sent you all the evidence that it is, in fact, completely reproducible (not "moderately" so as is stated on your website).
I understand that your blog is meant to be helpful but you should also remember (as Yong-Li, a 10 year Merck Process veteran reminded me the other day) that many reactions have a "window" of reproducibility. In the current case it might be with regards to the quality of the IBX used (your spectrum of "good" IBX does not look very good - see attached for an example of what good IBX looks like) and the actual internal temperature employed. If you are doing the reaction on something as volatile as toluene or 2-methylnapthalene then you must ensure that the entire flask, vial, or sealed tube is at 80-100 degrees or the SM will simply "hang out" at the the cooler part of the vessel. That's precisely why you and your colleagues saw little or no product because the SM was never actually exposed to the IBX (high or low quality it should have worked to some extent). Seems obvious in retrospect, and thats probably why it wasn't explicitly stated in the SI. That was our mistake.
Over the years my students have run dozens (if not hundreds) of literature reactions that either did not work in their hands or gave significantly lower yields of product than reported – before around 1995 many papers had no SI or procedures to speak of (except maybe JOC). I think all experienced organic chemists have seen this. That's why journals like Org. Syn. and Org. Proc. Res. Dev. exist. General procedures in an SI are mostly general guidelines. In the full paper we did give a number of examples of different conditions and it is up to the chemist to decide on the best ones for a given substrate (as the Kirsch group did in 2006 when they successfully oxidized several complex benzylic substrates at 110 degrees in DMSO with IBX).
Your blog will remind the community of the importance of documenting even the most minute and subtle details and pictures of reaction setups of new reactions. General procedures are clearly not always enough and, ideally, a Supporting Information should resemble an Organic Synthesis prep. Our group will certainly keep this in mind in the future and I suspect most of the academic community will too. And again, I am available day or night to anyone (anonymous or not) that has questions about anything I have co-authored.
Thanks for pushing the field in a positive direction.
Best Wishes,
phil
p.s. Tamsyn will also be emailing you again soon (you ignored her first email with suggestions) with her pictures of a reproduced reaction from a different lab.
––––––––––––––––––––––––––––––
The following is the procedure, TLC, NMRs (of IBX, 2-methylnapthalene, and product), and reaction setup used for 2-methylnapthalene (the substrate that you obtained "0% yield" on).
To a 1.25M solution of 2-methylnapthalene (distilled, 22 mg) in DMSO/PhF (5:1) was added IBX (NMR spectrum attached) and the reaction was heated to 95 degrees for 24 hours (TLC performed with 9:1 Hexanes:EtOAc). The vial was wrapped in foil and submerged as low as possible in the oil bath to prevent the volatile SM from escaping. As a control, if the reaction was done with pure DMSO and submerged only halfway, the napthalene refluxed at the top of the vial (confirmed by cooling it and watching the 2-methylnapthalene crystallize) and <10% conversion was observed since the reaction cannot take place if all reactants are not in solution. After 24 hours the reaction was worked up exactly as described in the SI (using 5% NaHCO3) and extracted with EtOAc. A silica column was run and 16mg of pristine product (65%) was obtained. I'm certain if this is done on larger scale and/or the solvent ratio is modified slightly a much higher yield can be obtained. This was essentially the first time (aside from the pure DMSO reaction) this student did this reaction and it will be the last."
 |
| SM |
 |
| SM in DMSO-d6 |
 |
| Fluorobenzene |
 |
| DMSO-d6 |
 |
| All SM before reaction. |
 |
| IBX in DMSO-d6 |
 |
| Rxn, showing 95 deg C |
 |
| Close up on oil bath w/vials |
 |
| TLC |
 |
| Final product |
 |
| Final pdt NMR, CDCl3 |
Author Addendum II: Dr. Tamsyn Montagnon has also written in with a duplicated reaction. Below, I've reproduced her email and pictures in their entirety:
"You may want to consider the following:
[1] Good IBX really is NOT hard to make. It should be free flowing, crystalline with a definite sparkle. See attached 1H NMR.
[2] There is in your discussion a throw away comment about the 2-methylnaphthalene not being volatile. Have you never showered and then left your hair to dry at room temperature? – water evaporates at RT, just as 2-methylnaphthalene does at 95 ºC. I suggest to you that at the working temperatures of this reaction, the biggest problem may be substrate evaporation (or “hanging out” on the colder parts of the flask as Phil said).
[3] As Phil also said every reaction has a window. I think we can all agree that the window for this reaction is pretty narrow and that what you are seeking in your blog-site work are wide windows, ones that aren’t so sensitive to the many variables in play. This, however, is a more complex discussion than you are making out to be.
[4] We need fluorobenzene to reflux in these reactions and in so-doing wash substrate back into the reaction from the walls of the vessel, but it clearly slows/inhibits the reaction also – so, we need to tune this balance carefully and with patience. Scale will have a big influence on how well this process works.
[5] Likewise, IBX decomposes under the reaction conditions (heating in a DMSO solution) so again we have to balance this with the need for higher temperatures to initiate the reaction when trying to find the best conditions for each substrate.
[6] I also believe the method used in isolation of the products is critical here. The reaction by-products are solids onto which the products stick (these hypervalent iodine compounds are all very oxaphilic Lewis acids). Without care, the layers are hard to separate and material gets lost in amongst the solid residue and poorly separated layers.
Moving on to the test reaction I conducted this week. Unfortunately, I work in a lab with extremely limited resources and therefore I have encountered some problems; the most important being we have no fluorobenzene on site, a lag time of a month on all orders we place for chemicals and very little money for consumables. For these reasons, I had to conduct the reaction with benzene instead of fluorobenzene – as you’ll appreciate I kept the scale small, because I really wasn’t keen on large scale benzene refluxing. It has to be said though that it is rather an important 5 ºC at the given reaction temperatures that we are talking about here (benzene refluxes at 80 ºC and fluorobenzene at 85 ºC), but despite this I obtained significant product (in contrast to your claim of no product). I also used 1-methylnapthalene not 2-methylnapthalene because this is all that we had.
Photos are attached of all relevant reagents/ set up etc.
Otherwise, I carried out the reaction exactly as Phil described in his procedure except that I added the IBX (3 equiv.) in four evenly spaced portions (be careful not to open up a hot reaction and loose all the substrate in the course of these additions though!). Also, with regard to the work up, I prefer to extract with diethyl ether because EtOAc or DCM drag IBX/by-product residues into the organic phase. I also tend to filter through a small pad of celite (after addition of diethyl ether) to remove solids.
I obtained 27 mg, 48 % of the desired aldehyde (1H NMR attached) and recovered 20 mg, 40 % unreacted start material."
 |
| Starting materials |
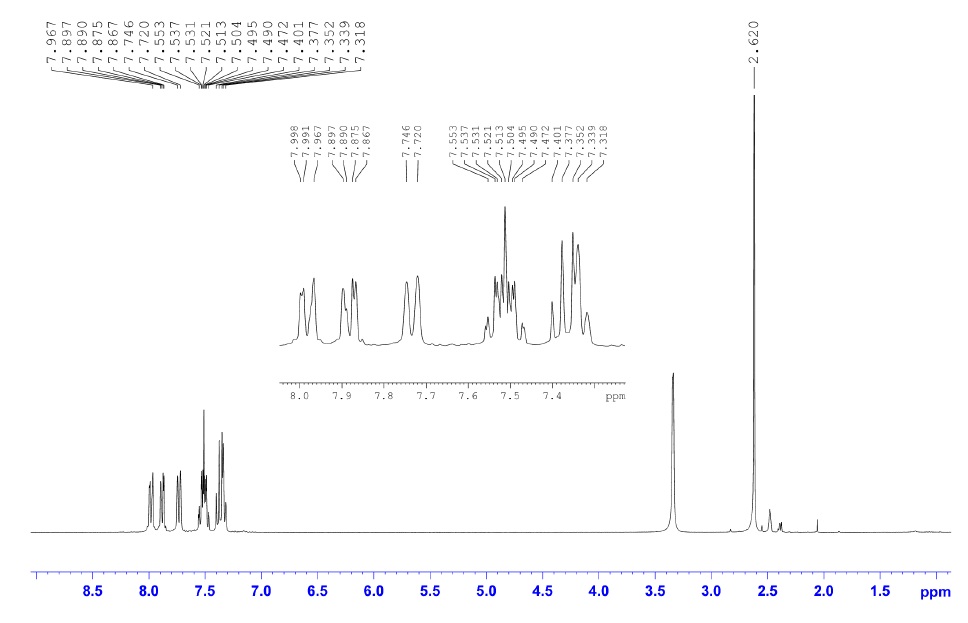 |
| 1-methylnaphthalene NMR |
 |
| IBX NMR |
 |
| Reaction setup |
 |
| TLC, crude reaction |
 |
| Pdt NMR, CDCl3 |
Roundup:
This reaction clearly requires mojo. We observe a fairly substantial difference between the authors’ results and ours; a quick poll around the blogosphere suggests others also have trouble with this reaction. An unclear Supporting Info, a highly-variable reagent, and possible physical phenomena (IBX solubility, temperature control, etc) could potentially explain our experimental results.
Given Blog Syn’s credo, we must deem this reaction Difficult to reproduce.
Thanks to B.R.S.M. (and friend) for synthetic runs, and for the second round based on Dr. Baran's advice. Readers, have you run this before, successfully? Please discuss your secrets in the Comments section.
Tuesday, February 12, 2013
Blog Syn #002: Pd-Catalyzed C-3 Selective C-H Olefination of Pyridines
(Disclaimer: The following experiments do not constitute rigorous peer review, but rather illustrate typical yields obtained and observations gleaned by trained synthetic chemists attempting to reproduce literature procedures. We've taken efforts to stay close to the original procedure, using similar glassware, equipment, and reagents wherever possible. Images have been cropped and scaled to fit in the allotted space, but have not been digitally altered otherwise.)
Updated: 2/20/13 - Added Yu rxn pictures
According to Web of Science, the article has been cited 45 times as of February 2013 (including 4 reviews). A cursory SciFinder search didn't find any examples with direct usage of this chemistry; an extension to arylation is found in a 2011 synthesis of preclamol by Yu's group.
Update(2-17): A related reaction for C-2 olefination of pyridines was reported in Adv. Synth. Cat. 2012, 354, 2135. We apologize for this omission.
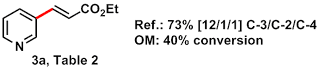 Scale: 0.5 mmol, as per SI conditions.
Scale: 0.5 mmol, as per SI conditions.
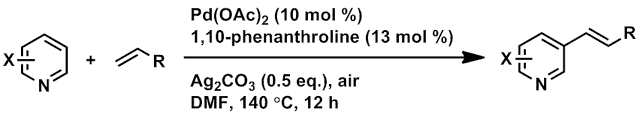
Entry: Compound 3a, Table 2 from JACS 2011, 6964.
Reagents: Palladium acetate [3375-31-3] (Strem, fresh bottle stored in glovebox), 1,10-phenanthroline [66-71-7] (Sigma-Aldrich, age unknown), Silver Carbonate [534-16-7] (Strem, ~2 mos old, stored in freezer in amber bottle within glovebox), ethyl acrylate [140-88-5] (Sigma, age unknown-purity >99% by NMR), Pyridine [110-86-1] (ACROS 99.8%), and DMF [68-12-2] (from solvent still, source unknown, see Advice for comments on this reagent).
Updated: 2/20/13 - Added Yu rxn pictures
Ref: Ye, M.; Gao, G.-L.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 6964-6967. doi: 10.1021/ja2021075.
Experimenters: Two - See Arr Oh (Somewhere, U.S.A.), Organometallica (IL)
Recommendation: Pending Blog Syn repeat
Initial - Moderately reproducible - yields/conversion lower than reported; reactivity / regioselectivity mostly consistent with literature. May require optimization.
Initial - Moderately reproducible - yields/conversion lower than reported; reactivity / regioselectivity mostly consistent with literature. May require optimization.
General scheme:
Context: This reaction comes from a 2011 paper (dx.doi.org/10.1021/ja2021075) by Jin-Quan Yu's group at the Scripps Research Institute titled "Ligand-Promoted C-3 Selective C- H Olefination of Pyridines with Pd Catalysts." See Arr Oh proposed this reaction, noting its potential to be "operationally simple...with few variables to goof up, and wide substrate availability." Synthetically, C-H activation appeals to synthetic chemists due to its high atom efficiency (less waste) and simplicity (no need to pre-functionalize a reaction center).
The procedure employs a Pd catalyst to 'activate' the pyridyl C-3 carbon-hydrogen bond. A bidentate pyridyl ligand, phenanthroline (phen), presumably enhances ligand exchange via the trans-effect (to counteract the strong coordination of the pyridine substrate to the metal center). The reaction is run with a Ag(I) co-catalyst under air, which serves as the terminal oxidant.
The authors highlight the important of 3-alkenyl pyridine derivatives in chemistry, citing numerous natural products and pharmaceuticals exhibiting this motif. They claim the first instance of C-3 selective pyridine olefination (a number of examples with more limited substrate scope and different site selectivity are given). Just how C-3 selective? Yu and coworkers report C-3/C-2/C-4 site selectivity ranging from 7/1/1 to 30/1/1.
The procedure employs a Pd catalyst to 'activate' the pyridyl C-3 carbon-hydrogen bond. A bidentate pyridyl ligand, phenanthroline (phen), presumably enhances ligand exchange via the trans-effect (to counteract the strong coordination of the pyridine substrate to the metal center). The reaction is run with a Ag(I) co-catalyst under air, which serves as the terminal oxidant.
The authors highlight the important of 3-alkenyl pyridine derivatives in chemistry, citing numerous natural products and pharmaceuticals exhibiting this motif. They claim the first instance of C-3 selective pyridine olefination (a number of examples with more limited substrate scope and different site selectivity are given). Just how C-3 selective? Yu and coworkers report C-3/C-2/C-4 site selectivity ranging from 7/1/1 to 30/1/1.
According to Web of Science, the article has been cited 45 times as of February 2013 (including 4 reviews). A cursory SciFinder search didn't find any examples with direct usage of this chemistry; an extension to arylation is found in a 2011 synthesis of preclamol by Yu's group.
Update(2-17): A related reaction for C-2 olefination of pyridines was reported in Adv. Synth. Cat. 2012, 354, 2135. We apologize for this omission.
Experimental
Trial 1 - See Arr Oh
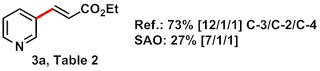 Scale: 0.5 mmol (same as researchers)
Scale: 0.5 mmol (same as researchers)
Entry: Compound 3a, Table 2 from JACS 2011, 6964.
Reagents: ACROS Ethyl acrylate [140-88-5], 99.5%; Alfa silver carbonate [534-16-7], 99.5%; ALD anhydrous pyridine [110-86-1], 99.8%; STREM Pd(OAc)2 [3375-31-3], 99%; ALD 1,10-phen [66-71-7], Aldrich DMF, anh. 99.8%
Glassware: Pressure tube (75 mL, Teflon bushing) oven-dried o/n. All dry reagents weighed out in air.
Observations: First 5 mins: yellow pasty suspension. At temp (141.3 deg, sand bath) - deep orange-brown. After 16 h - deep brown, almost homogeneous, silvery Ag(0) / Pd(0) sheen around top of flask. Diluted with EtOAc, filtered over 1/2" Celite plug (2 x 5mL wash), then column chromatography (3 x 1 cm, silica, 1:4 - 1:1 EA / Hept).
Pale yellow oil, 7/1/1 meta / ortho / para, 27% yield overall (38 mg isol'd: 24 mg pdt + 14 mg DMF by NMR integration)
Advice: Crude product an orange gum, tough to handle / redissolve. I would re-load onto silica, and incorporate a water wash to eliminate DMF.
Supplementary images (click to enlarge):
Trials 2 and 3 - Organometallica
Entry: Compound 3a, Table 2 from JACS 2011, 6964.
 |
| Reaction in 75 mL sealed pressure tube. |
Glassware: Pressure tube (75 mL, Teflon bushing) oven-dried o/n. All dry reagents weighed out in air.
Observations: First 5 mins: yellow pasty suspension. At temp (141.3 deg, sand bath) - deep orange-brown. After 16 h - deep brown, almost homogeneous, silvery Ag(0) / Pd(0) sheen around top of flask. Diluted with EtOAc, filtered over 1/2" Celite plug (2 x 5mL wash), then column chromatography (3 x 1 cm, silica, 1:4 - 1:1 EA / Hept).
Pale yellow oil, 7/1/1 meta / ortho / para, 27% yield overall (38 mg isol'd: 24 mg pdt + 14 mg DMF by NMR integration)
Advice: Crude product an orange gum, tough to handle / redissolve. I would re-load onto silica, and incorporate a water wash to eliminate DMF.
Supplementary images (click to enlarge):
 |
| 1H NMR spectrum, post-chromatography. |
 |
| Reaction setup, before heating. |
 |
| TLC: left, ethyl acrylate center, co-spot right, reaction mixture |
 |
| Filter. |
 |
| Reagents employed. |
 |
| Measured reagents. |
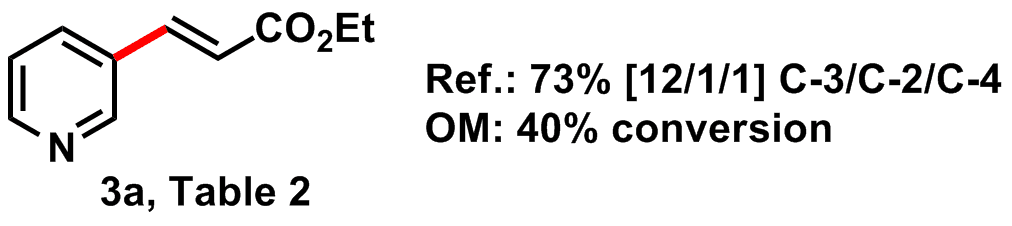
Entry: Compound 3a, Table 2 from JACS 2011, 6964.
Reagents: Palladium acetate [3375-31-3] (Strem, fresh bottle stored in glovebox), 1,10-phenanthroline [66-71-7] (Sigma-Aldrich, age unknown), Silver Carbonate [534-16-7] (Strem, ~2 mos old, stored in freezer in amber bottle within glovebox), ethyl acrylate [140-88-5] (Sigma, age unknown-purity >99% by NMR), Pyridine [110-86-1] (ACROS 99.8%), and DMF [68-12-2] (from solvent still, source unknown, see Advice for comments on this reagent).
Glassware: Pressure tube (75 mL).
Observations: NOTE: This reaction was attempted multiple times. In all cases, the reaction was set up on the bench without precautions against preventing air contamination. Reagents were added neat to 75 mL pressure vessel in the order Pd(OAc)2, 1,10-phenanthroline, Ag2CO3, pyridine, ethyl acrylate, followed by DMF, then sealed and heated at 140˚C for 12 hrs.
For Trial 2, DMF used directly from solvent still, for Trial 3, air was bubbled through DMF for 1 hour prior to use. Trial 2: Reaction darkens, remains heterogeneous. Trial 3 (Pictured): Turns pasty yellow, still some solid left in the flask.
Analysis: 1H NMR: Trial 2: no conversion observed. Trial 3: ~40% conversion.
Advice: When I first ran this reaction (Trial 2), I panicked. There was absolutely no product; nothing by TLC, nothing by NMR, nothing by GC-MS. However, one of my friends who work on Pd-catalyzed C-H activation remarked that reactions run under air are particularly sensitive to solvent O2 concentration, so much so that he saturates solvents with O2 before use. Thinking this might be what plagued me, I aerated my solvent (Trial 3), which seemed to kick off the reaction. This may be the first time I've ever had a reaction fail for having my reagents be too clean!
Supplementary images (click to enlarge):
Thanks to See Arr Oh and Organometallica for experiments, and thanks to Prof. Yu for providing feedback. Readers: if you have experience with this reaction or suggestions for future investigations, please feel free to comment.
*Addendum (Feb 20, 2013): After an exchange of emails, Prof. Yu felt it best to have a new postdoc (not an original paper author) repeat the experiment, and sent over pictures of the reaction setup, along with a crude NMR.


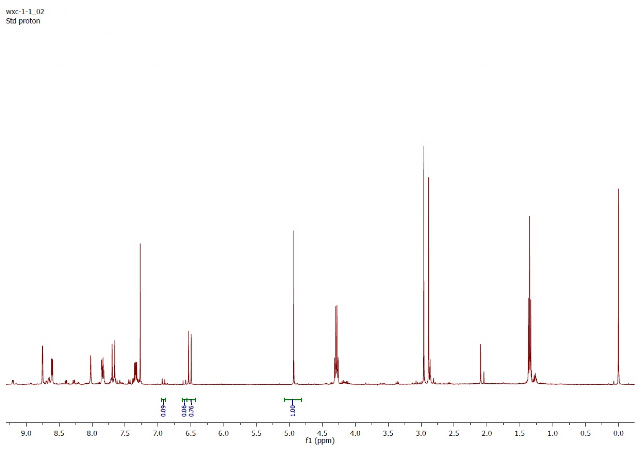
The ratio I get from these integrations is 12.7 / 1.5 / 1 (m/o/p), which agrees with the original publication.
Update (2/20/13): Prof. Yu indicates that integration of the crude against 0.5 equiv. of CH2Br2 (4.94 ppm, above) gives an NMR yield of meta-olefination product matching the literature (76%).
Of note, the reagents are quite different - he's using a higher grade of both (1,10-phen) and pyridine, but we don't have information on the acrylate used or the Pd source. Also of note, the 50 mL long, cylindrical tube volume clearly benefits the reaction, as alluded to in Prof. Yu's original email.
We thank Prof. Yu for sending along some clarification. Perhaps this signifies a change in the way reactions are reported; if everyone had to send along photo evidence, would there be so many reproducibility issues in the modern literature?
-SAO
 |
| Trial 3; reaction setup. |
For Trial 2, DMF used directly from solvent still, for Trial 3, air was bubbled through DMF for 1 hour prior to use. Trial 2: Reaction darkens, remains heterogeneous. Trial 3 (Pictured): Turns pasty yellow, still some solid left in the flask.
Analysis: 1H NMR: Trial 2: no conversion observed. Trial 3: ~40% conversion.
Advice: When I first ran this reaction (Trial 2), I panicked. There was absolutely no product; nothing by TLC, nothing by NMR, nothing by GC-MS. However, one of my friends who work on Pd-catalyzed C-H activation remarked that reactions run under air are particularly sensitive to solvent O2 concentration, so much so that he saturates solvents with O2 before use. Thinking this might be what plagued me, I aerated my solvent (Trial 3), which seemed to kick off the reaction. This may be the first time I've ever had a reaction fail for having my reagents be too clean!
Supplementary images (click to enlarge):
 |
| 1H NMR spectrum, full |
 |
| 1H NMR spectrum, expanded to show aromatic region |
Results and Discussion
Author* response: Corresponding author Jin-Quan Yu (Scripps) was kind enough to respond with some helpful advice (Bold highlights are mine).
1. As what you have observed, the oxygen concentration is very important to this reaction. So the tube volume is also very important. We used the 50-mL tube, which is the total volume of the tube (from the bottom to the cap). For smaller one, the air is not enough. For bigger one, the pyridine will volatilize too much outside the solution.
2. Heating and stirring is another important thing. Prior to starting experiments, turn on the hot plate and set the oil bath to 140°C first. To make the oil temperature is stable, you can put a paper clip into the oil, which will help to stir the oil. Put the reaction tube in the middle of hot plate to obtain a stable stirring (500 rpm is enough). Don't let the solid adhere to the tube wall. And make sure the surface of reaction solution is lower than oil surface.
3. Another thing I should mention is that the quality of Ag2CO3 and Phen may ruin this reaction. We found that these chemicals from Strem Chemicals are good quality and there is no risk for this reaction.
Roundup: The reaction certainly works, though certain variables essential to its success are under-emphasized in the original paper (see Author response) and were unfortunately omitted from the SI. Notably, conversion depends on the presence of oxygen in solution (see Organometallica's trials). Even solvent aeration produced substantially lower conversion than the authors report (40% vs. 73% isolated yield). See Arr Oh did, however, observe similar regioselectivity (7/1/1 vs. 12/1/1).
Since both yields and observed regioselectivities fall within the low end of those reported, we classify this procedure as moderately reproducible.
Since both yields and observed regioselectivities fall within the low end of those reported, we classify this procedure as moderately reproducible.
Thanks to See Arr Oh and Organometallica for experiments, and thanks to Prof. Yu for providing feedback. Readers: if you have experience with this reaction or suggestions for future investigations, please feel free to comment.
*Addendum (Feb 20, 2013): After an exchange of emails, Prof. Yu felt it best to have a new postdoc (not an original paper author) repeat the experiment, and sent over pictures of the reaction setup, along with a crude NMR.



The ratio I get from these integrations is 12.7 / 1.5 / 1 (m/o/p), which agrees with the original publication.
Update (2/20/13): Prof. Yu indicates that integration of the crude against 0.5 equiv. of CH2Br2 (4.94 ppm, above) gives an NMR yield of meta-olefination product matching the literature (76%).
Of note, the reagents are quite different - he's using a higher grade of both (1,10-phen) and pyridine, but we don't have information on the acrylate used or the Pd source. Also of note, the 50 mL long, cylindrical tube volume clearly benefits the reaction, as alluded to in Prof. Yu's original email.
We thank Prof. Yu for sending along some clarification. Perhaps this signifies a change in the way reactions are reported; if everyone had to send along photo evidence, would there be so many reproducibility issues in the modern literature?
-SAO
Wednesday, January 16, 2013
Blog Syn #001 - Iron / Sulfur Catalysis
(Disclaimer: The following experiments do not constitute rigorous peer review, but rather illustrate typical yields obtained and observations gleaned by trained synthetic chemists attempting to reproduce literature procedures. We've taken efforts to stay close to the original procedure, using similar glassware, equipment, and reagents wherever possible. Images have been cropped and scaled to fit in the allotted space, but have not been digitally altered otherwise.)
Ref: Nguyen, T.B.; Ermolenko, L.; Al-Mourabit, A. J. Am. Chem. Soc. 2013, 135(1), 118-121.
Previous Analysis: Just Like Cooking: Post 1, Post 2, author response
Experimenters: Three - Matt Katcher (NJ), Organometallica (IL), B.R.S.M. (United Kingdom)
Recommendation: Moderately reproducible - yields much lower than anticipated
 Trial 1 - Matt
Trial 1 - Matt
Scale: 1 mmol
Reaction (table, #): Table 2, entry 2
 Trial 2 - B.R.S.M.
Trial 2 - B.R.S.M.
Scale: 5 mmol
Reaction (table, #): Table 2, entry 1
Reagents: 2-Nitroaniline (Aldrich 98%; of indeterminate age, because I borrowed it), sulfur (Fisher 'lab reagent' grade, fine powder, age unknown), iron (Fisher '97% reduced by hydrogen grade' [no mesh given]), 4-picoline (10 mmol; Aldrich 98%; 1 year old)
Observations: Used 10-mL Schlenk tube that had been dried under vacuum using a heat-gun. The flask was evacuated and back-filled with argon three times. Dark-coloured suspension, agitated at maximum speed by magnetic stir bar.
Purification: Silica gel. Elution with 100:0 - 95:5 CH2Cl2:MeOH gave the product as a beige foam, still containing some impurities (Rf 0.45 in 96:4). Second attempt: Recrystallisation from toluene-hexane (ca 3:1) gave small yellow-golden brown crystals that were dried overnight in vacuo.
Yield - Published: 90%
Actual: before recryst 366 mg (37%); after 241 mg (24%).
Advice: A crude sample of reaction mixture removed at 24 h indicated around 5:1 aniline:product, but this was probably not reliable due to the low solubility of the product in chloroform. The reaction was quite messy, the product streaky and the column non-trivial.
Spectra / Photos:
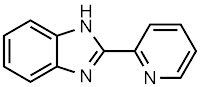 Trial 3 - Organometallica
Trial 3 - Organometallica
Scale: 5 mmol
Reaction (table, #): Table 2, entry 3
Reagents: Fe (Aldrich, >99% powder, fine), S (Aldrich, >99.998%, recrystallized)
Observations: Reaction run in inert-atmosphere glovebox in Schlenk bomb. Noted "internet accounts" (trials 1 & 2) before beginning this reaction.
Purification: Pushed through silica pad with DCM, 10%MeOH / DCM. Distilled on bulb-to-bulb apparatus to remove excess picoline.
Yield - Published: 75%
Actual: 35%
Advice: No residual product in picoline distillate or remaining on silica plug. Flask / reaction capable of withstanding increased pressure. Deep brown crude mixture, final product a green powder.
Spectra / Photos:
Roundup: First, a hearty thanks to all three experimenters for taking time out of their busy schedules to run these reactions. Second, I believe we'd all agree that the reaction works, but in practice we obtain far less material than the original authors. Oxygen contamination plays a major role - note lower yield in Trial 1 - as (we suspect) does mesh size and age of iron and sulfur powders.
Author Response: (When this post goes live, I'll send the link to the corresponding author. Any response will be published in due course)
**Thanks for reading our initial venture into collaborative "crowdsourced reaction validation." We appreciate any comments or suggestions for the next go-around. Want to get involved? Have an idea for another (cheap!) reaction to try? Talk to us in the Comments section!
Ref: Nguyen, T.B.; Ermolenko, L.; Al-Mourabit, A. J. Am. Chem. Soc. 2013, 135(1), 118-121.
Previous Analysis: Just Like Cooking: Post 1, Post 2, author response
Experimenters: Three - Matt Katcher (NJ), Organometallica (IL), B.R.S.M. (United Kingdom)
Recommendation: Moderately reproducible - yields much lower than anticipated
Trial 1 - MattScale: 1 mmol
Reaction (table, #): Table 2, entry 2
Reagents: Fe was an old Fisher bottle (40 mesh)
Observations: Tried to keep reaction air-free by setting up with a septum over a pressure tube, but I was not extremely rigorous. Fe stuck to stir bar at end of reaction
Purification: Not attempted
Yield - Published: 83%
Actual: <5% by crude NMR
Advice: I still think the reaction might work, but it may not be a simple "dump and stir". If I did it again, I would run it in a Schlenk tube to minimize exposure to air.
Spectra / Photos:
 |
| Reaction setup |
 |
| LC/MS - Target ion: 246 m/z |
 |
| TLC |
 |
| Crude NMR |
Trial 2 - B.R.S.M.Scale: 5 mmol
Reaction (table, #): Table 2, entry 1
Reagents: 2-Nitroaniline (Aldrich 98%; of indeterminate age, because I borrowed it), sulfur (Fisher 'lab reagent' grade, fine powder, age unknown), iron (Fisher '97% reduced by hydrogen grade' [no mesh given]), 4-picoline (10 mmol; Aldrich 98%; 1 year old)
Observations: Used 10-mL Schlenk tube that had been dried under vacuum using a heat-gun. The flask was evacuated and back-filled with argon three times. Dark-coloured suspension, agitated at maximum speed by magnetic stir bar.
Purification: Silica gel. Elution with 100:0 - 95:5 CH2Cl2:MeOH gave the product as a beige foam, still containing some impurities (Rf 0.45 in 96:4). Second attempt: Recrystallisation from toluene-hexane (ca 3:1) gave small yellow-golden brown crystals that were dried overnight in vacuo.
Yield - Published: 90%
Actual: before recryst 366 mg (37%); after 241 mg (24%).
Advice: A crude sample of reaction mixture removed at 24 h indicated around 5:1 aniline:product, but this was probably not reliable due to the low solubility of the product in chloroform. The reaction was quite messy, the product streaky and the column non-trivial.
Spectra / Photos:
 |
| 400 MHz NMR, after rexstal |
 |
| Final product |
Trial 3 - OrganometallicaScale: 5 mmol
Reaction (table, #): Table 2, entry 3
Reagents: Fe (Aldrich, >99% powder, fine), S (Aldrich, >99.998%, recrystallized)
Observations: Reaction run in inert-atmosphere glovebox in Schlenk bomb. Noted "internet accounts" (trials 1 & 2) before beginning this reaction.
Purification: Pushed through silica pad with DCM, 10%MeOH / DCM. Distilled on bulb-to-bulb apparatus to remove excess picoline.
Yield - Published: 75%
Actual: 35%
Advice: No residual product in picoline distillate or remaining on silica plug. Flask / reaction capable of withstanding increased pressure. Deep brown crude mixture, final product a green powder.
Spectra / Photos:
 |
| NMR, post-distillation |
 |
| TLC, from notebook |
Author Response: (When this post goes live, I'll send the link to the corresponding author. Any response will be published in due course)
**Thanks for reading our initial venture into collaborative "crowdsourced reaction validation." We appreciate any comments or suggestions for the next go-around. Want to get involved? Have an idea for another (cheap!) reaction to try? Talk to us in the Comments section!
Subscribe to:
Posts (Atom)